




合作客戶/
拜耳公司 |
同濟大學 |
聯合大學 |
美國保潔 |
美國強生 |
瑞士羅氏 |






相關新聞Info
超低界面張力環保型高溫高鹽油藏的驅油表面活性劑配方比例及制備(二)
來源:匯通路橋集團試驗檢測有限公司 保定職業技術學院 瀏覽 841 次 發布時間:2025-11-14
實施例1
一種環保型表面活性劑組合物,按重量份數計,包括以下組分:陽離子表面活性劑58份、乳化劑42份(蓖麻油聚氧乙烯醚EL-40)、N,N-二羥乙基甘氨酸2份、溶劑(水)23份;
所述陽離子表面活性劑的制備方法如下:
(1)將維生素E、碳酸鉀、碘化鉀加入至無水N,N-二甲基甲酰胺,然后在室溫下滴加2-溴乙基丙烯酸酯,其中,維生素E、碳酸鉀、碘化鉀、2-溴乙基丙烯酸酯的摩爾比為1:2.2:0.3:1.6;所述維生素E和無水N,N-二甲基甲酰胺的用量比為10mmol:55mL,攪拌均勻后升溫至55℃反應2.5h;隨后將混合物冷卻至室溫,用乙酸乙酯萃取,合并有機層,依次用5%氫氧化鈉水溶液和水洗滌、無水硫酸鈉干燥、過濾、濃縮后即得中間體1;HRMS-ESI(m/z):[M+H]+calcd for C34H56O4,529.42;found,529.48.
(2)將N-芐基D-葡糖胺、中間體1和三乙胺加入至甲苯中,所述中間體1、N-芐基D-葡糖胺和三乙胺的摩爾比為5:7:6.5;中間體1和甲苯的用量比為5mmol:55mL,于室溫下反應14h;濃縮反應液后經硅膠柱層析(石油醚/乙酸乙酯=5:1)純化,即得中間體2;1H NMR(400 MHz,DMSO-d6):7.30-7.22(m,5H),4.50-4.38(m,8H),3.95(s,1H),3.62-3.41(m,8H),2.85-2.75(m,4H),2.62-2.41(m,4H),2.08(s,9H),2.01-1.81(m,2H),1.62-1.57(m,3H),1.42-1.40(m,5H),1.26-1.20(m,16H),0.90(t,12H);HRMS-ESI(m/z):[M+H]+calcd for C47H77NO9,800.56;found,800.56.
(3)將中間體2置于二氯甲烷中,加入碘甲烷,其中,中間體2、碘甲烷的摩爾比為1:3.5;所述中間體2和二氯甲烷的用量比為5mmol:55mL,于常溫下反應28h;濃縮反應液后經蒸餾純化,即得陽離子表面活性劑;1H NMR(400 MHz,DMSO-d6):7.25-7.17(m,5H),4.50-4.38(m,10H),4.14-4.12(m,1H),3.95(s,1H),3.63-3.50(m,6H),3.43-3.41(m,2H),3.35(s,3H),3.25-3.23(m,1H),2.85-2.69(m,4H),2.08(s,9H),2.01-1.81(m,2H),1.62-1.57(m,3H),1.42-1.40(m,5H),1.26-1.20(m,16H),0.90(t,12H);HRMS-ESI(m/z):[M]calcd for C48H80NO9,814.58;found,814.58.
本實施例還提供環保型表面活性劑組合物的制備過程為:將陽離子表面活性劑、乳化劑、N,N-二羥乙基甘氨酸、溶劑混合均勻即可。
實施例2
一種環保型表面活性劑組合物,按重量份數計,包括以下組分:陽離子表面活性劑55份、乳化劑40份(蓖麻油聚氧乙烯醚EL-40)、N,N-二羥乙基甘氨酸1份、溶劑20份(乙醇);
所述陽離子表面活性劑的制備方法如下:
(1)將維生素E、碳酸鉀、碘化鉀加入至無水N,N-二甲基甲酰胺,然后在室溫下滴加2-溴乙基丙烯酸酯,其中,維生素E、碳酸鉀、碘化鉀、2-溴乙基丙烯酸酯的摩爾比為1:2:0.2:1.5;所述維生素E和無水N,N-二甲基甲酰胺的用量比為10mmol:50mL,攪拌均勻后升溫至50℃反應3h;隨后將混合物冷卻至室溫,用乙酸乙酯萃取,合并有機層,依次用5%氫氧化鈉水溶液和水洗滌、無水硫酸鈉干燥、過濾、濃縮后即得中間體1;HRMS-ESI的表征結果同實施例1;
(2)將N-芐基D-葡糖胺、中間體1和三乙胺加入至甲苯中,所述中間體1、N-芐基D-葡糖胺和三乙胺的摩爾比為5:5:5.5;中間體1和甲苯的用量比為5mmol:50mL,于室溫下反應12h;濃縮反應液后經硅膠柱層析(石油醚/乙酸乙酯=5:1)純化,即得中間體2;1
H NMR和HRMS-ESI的表征結果同實施例1;
(3)將中間體2置于二氯甲烷中,加入碘甲烷,其中,中間體2、碘甲烷的摩爾比為1:3;所述中間體2和二氯甲烷的用量比為5mmol:50mL,于常溫下反應24h;濃縮反應液后經蒸餾純化,即得陽離子表面活性劑;1
H NMR和HRMS-ESI表征結果同實施例1。
本實施例還提供環保型表面活性劑組合物的制備過程為:將陽離子表面活性劑、乳化劑、N,N-二羥乙基甘氨酸、溶劑混合均勻即可。
實施例3
一種環保型表面活性劑組合物,按重量份數計,包括以下組分:陽離子表面活性劑60份、乳化劑45份(蓖麻油聚氧乙烯醚EL-40)、N,N-二羥乙基甘氨酸3份、溶劑25份(水);
所述陽離子表面活性劑的制備方法如下:
(1)將維生素E、碳酸鉀、碘化鉀加入至無水N,N-二甲基甲酰胺,然后在室溫下滴加2-溴乙基丙烯酸酯,其中,維生素E、碳酸鉀、碘化鉀、2-溴乙基丙烯酸酯的摩爾比為1:2.5:0.5:1.8;所述維生素E和無水N,N-二甲基甲酰胺的用量比為10mmol:60mL,攪拌均勻后升溫至60℃反應2h;隨后將混合物冷卻至室溫,用乙酸乙酯萃取,合并有機層,依次用5%氫氧化鈉水溶液和水洗滌、無水硫酸鈉干燥、過濾、濃縮后即得中間體1;HRMS-ESI的表征結果同實施例1;
(2)將N-芐基D-葡糖胺、中間體1和三乙胺加入至甲苯中,所述中間體1、N-芐基D-葡糖胺和三乙胺的摩爾比為5:8:8;中間體1和甲苯的用量比為5mmol:60mL,于室溫下反應16h;濃縮反應液后經硅膠柱層析(石油醚/乙酸乙酯=5:1)純化,即得中間體2;1
H NMR和HRMS-ESI的表征結果同實施例1;
(3)將中間體2置于二氯甲烷中,加入碘甲烷,其中,中間體2、碘甲烷的摩爾比為1:4;所述中間體2和二氯甲烷的用量比為5mmol:60mL,于常溫下反應36h;濃縮反應液后經蒸餾純化,即得陽離子表面活性劑;1
H NMR和HRMS-ESI表征結果同實施例1。
本實施例還提供環保型表面活性劑組合物的制備過程為:將陽離子表面活性劑、乳化劑、N,N-二羥乙基甘氨酸、溶劑混合均勻即可。
對比例1
對比例1和實施例1的區別在于:將陽離子表面活性劑替換為維生素E,其它和實施例1保持一致。
對比例2
對比例2和實施例1的區別在于:省去N,N-二羥乙基甘氨酸,其它和實施例1保持一致。
試驗例1
1、抗鹽性測試:配制高鹽模擬鹽水:礦化度32000mg/L,Ca2+、+Mg2+為1500mg/L;收集上述實施例1-3以及對比例1-2中制得的表面活性劑組合物,采用高鹽模擬鹽水將其配置成質量濃度為0.3%的表面活性劑組合物溶液,根據《SYT 5755-2016壓裂酸化用助排劑性能評價方法》中的方法測量溶液與原油(黏度為12mPa·s,溫度為45℃)之間的界面張力,測量溫度為25℃。
2、熱穩定性測試:收集上述實施例1-3以及對比例1-2中制得的表面活性劑組合物,采用高鹽模擬鹽水(礦化度32000mg/L,Ca2+
+Mg2+
為1500mg/L)將其配制成質量濃度為0.3%的水溶液,在80℃條件下放置20天后取出根據《SYT 5755-2016壓裂酸化用助排劑性能評價方法》中的方法檢測其界面張力,以界面張力評價樣品的熱穩定性。
上述測試結果記錄在表1中。
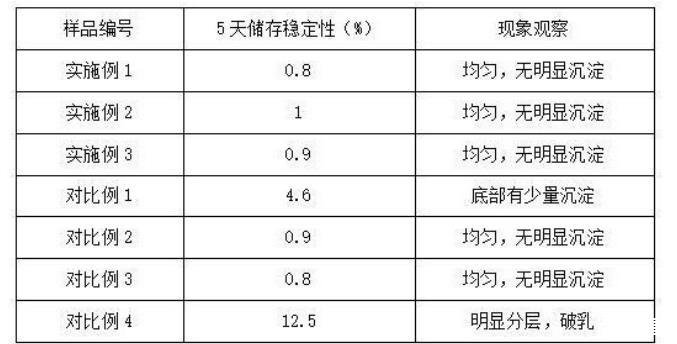
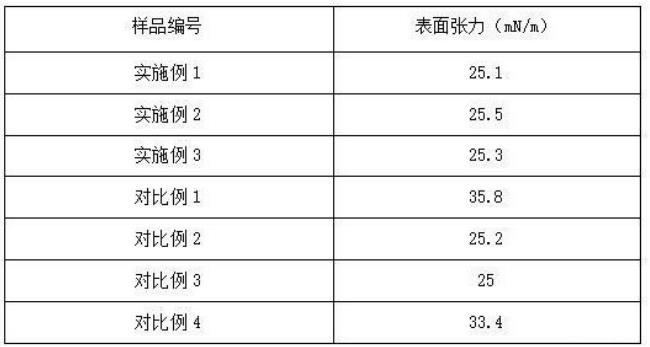
由表1的測試結果可知,本發明實施例1-3所得表面活性劑組合物具有良好的界面張力,并且熱穩定性和抗鹽性良好。相較于實施例1,對比例1將陽離子表面活性劑替換為維生素E,對比例2省去N,N-二羥乙基甘氨酸,二者的界面張力均下降。說明本發明所得陽離子表面活性劑在提升表面活性劑的熱穩定性和抗鹽性方面具有重要作用,并且能夠和N,N-二羥乙基甘氨酸二者協同作用,穩定陽離子表面活性劑的正電荷形態,促進油-水乳化,并降低界面張力。
試驗例2收集實施例1-3和對比例1-2所得表面活性劑組合物,分別采用礦化度32000mg/L,Ca2+、+Mg2+為1500mg/L的鹽水制成濃度為0.3wt%的表面活性劑組合物溶液;采用SY/T 6424-2014復合驅油體系性能測試方法模擬驅油效果測試。
實驗步驟為:①采用礦化度32000mg/L,Ca2+、+Mg2+為1500mg/L的注入水將巖心(長度為30厘米,直徑為2.5厘米,滲透率為1.5μm2
)飽和,測量飽和水量,計算巖心孔隙體積(PV);②恒溫箱加熱至60℃,老化巖心24 h;③將巖心以原油(溫度45℃時黏度為12mPa·s)進行飽和,并計算飽和油量及含油飽和度,然后將巖心靜置在地層溫度(60℃)環境中;④注入水進行恒速水驅,水驅至含水98%;計算水驅采收率;⑤水驅結束后,轉注0.5PV(巖心孔隙體積)的實施例1-3和對比例1和2所得表面活性劑組合物溶液,然后水驅至含水99%,計算注入表面活性劑組合物水驅后提高原油采收率。
由表2的測試結果可知,本發明采用實施例1-3的表面活性劑組合物進行水驅后提高采收率均為23%以上,明顯高于對比例1和2。這一結果說明本發明表面活性劑組合物中的陽離子表面活性劑和N,N-二羥乙基甘氨酸能夠共同作用,提高原油的采收率。